
Hi all,
I'm a PhD student in #MachineLearning at the technical university of Munich #TUM. I'm currently working on machine learning on graphs and machine learning-driven computional chemistry.
#ml #GraphNeuralNetworks #GNNs #compchem

@nicholasgao Hey Nicholas, what are you making with these Machine learning of chemical graphs?
Is it some kind of qsar?
@rastinza
Until now, my work has mostly been focused on machine learning potentials or ab-initio quantum chemistry, e.g.,
https://openreview.net/forum?id=apv504XsysP
@nicholasgao The idea looks very cool! Does it work?
@nicholasgao Sorry, I did not read the article at all.
What do you mean by not sacrificing performance? You get results comparable to ab initio calculations? What order of approximation are we talking about?
@nicholasgao Wow, that's really impressive! Such small errors are not what I was expecting.
Can this be applied to larger systems?
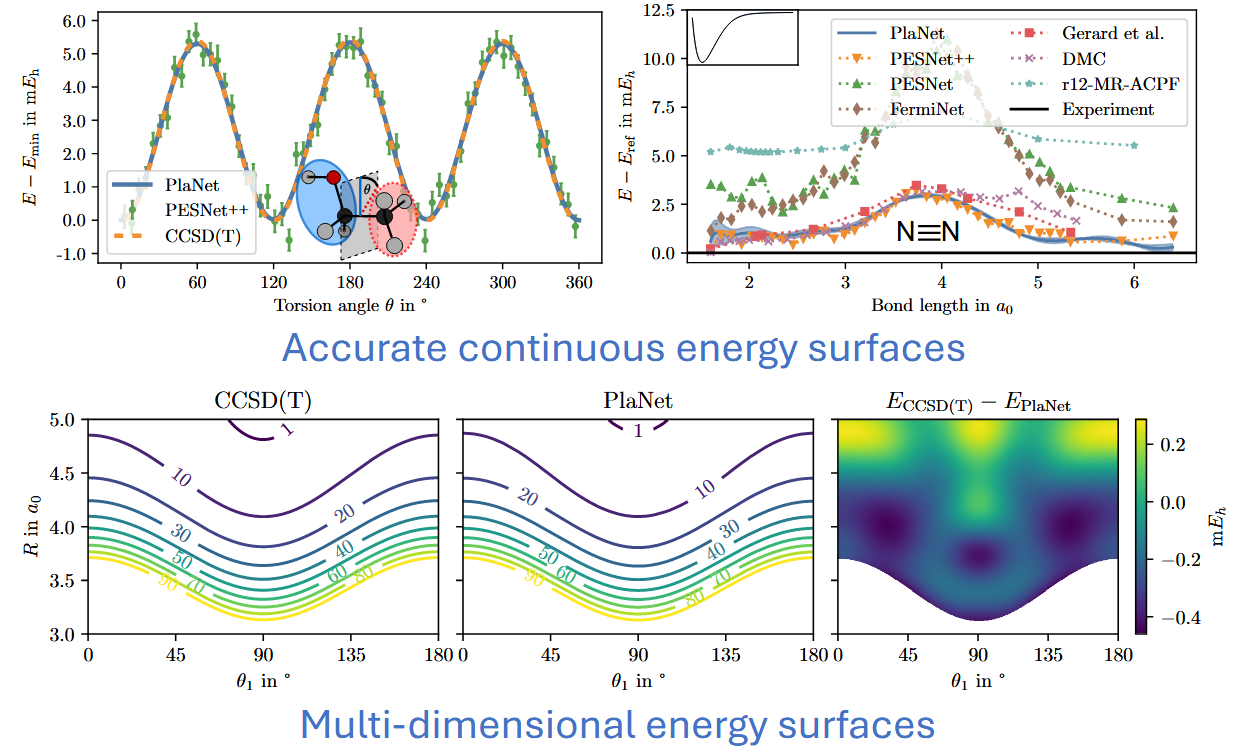
@rastinza In a recent preprint (https://arxiv.org/abs/2205.14962), we have shown that one can also get obtain continuous energy surfaces (denotes as PlaNet) for multi-dimensional data and "larger" molecules such as ethanol. Unfortunately, for systems larger than 40 electrons, these methods get quite expensive. So, their scaling in terms of size is still very much active research.
{kind=link}

@rastinza we in fact perform highly accurate ab-initio calculations. In many small system, such ML-driven ab-initio methods report the lowest variational results in the literature. What we mean with "not sacrificing performance" is that compared to neural wave function-based baselines we don't lose any accuracy despite solving many Schrödinger equations simultaneously.
The plot below shows our Potential Energy Surface Network (PESNet) in comparison to other neural wave function-based methods.